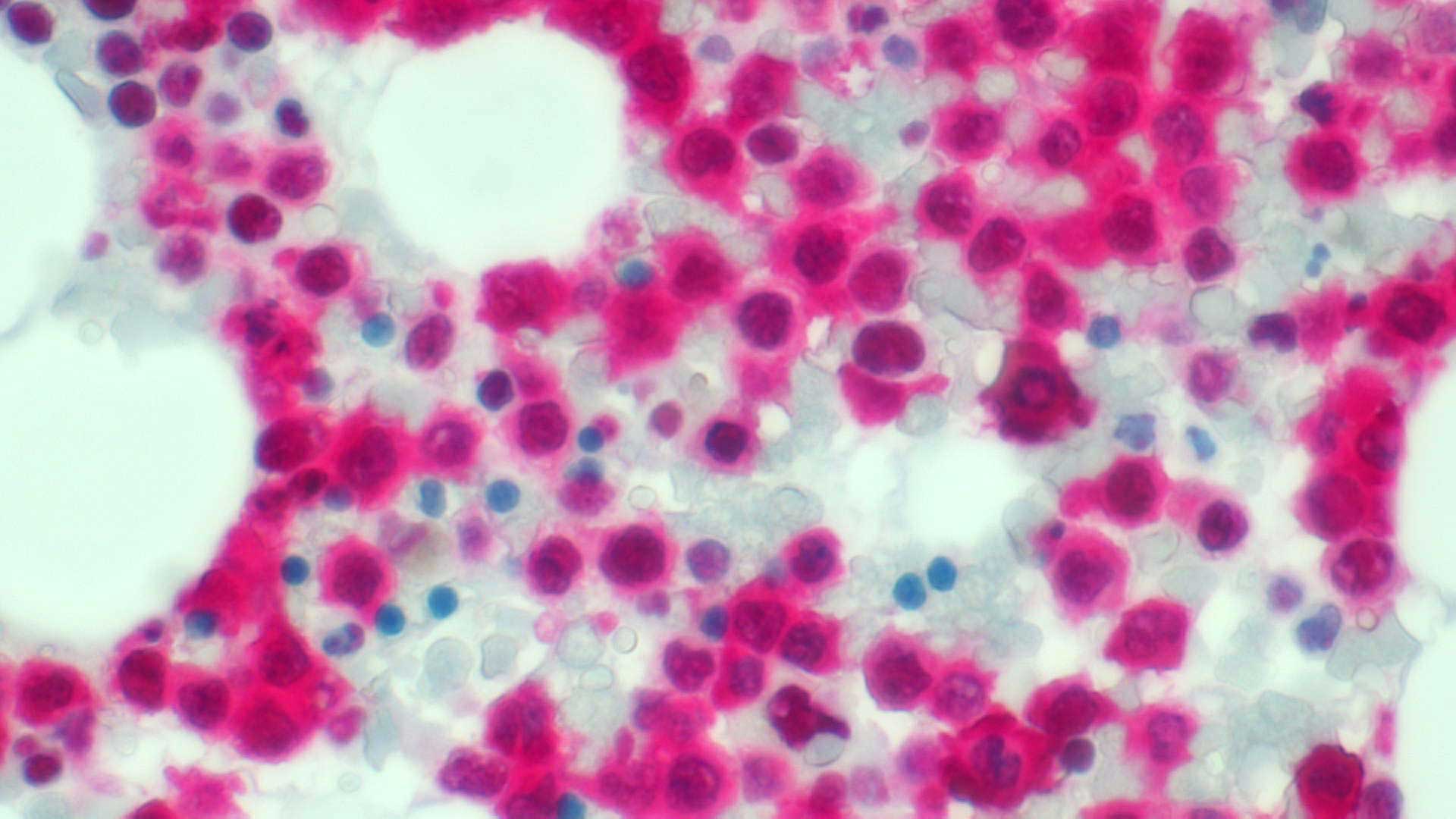
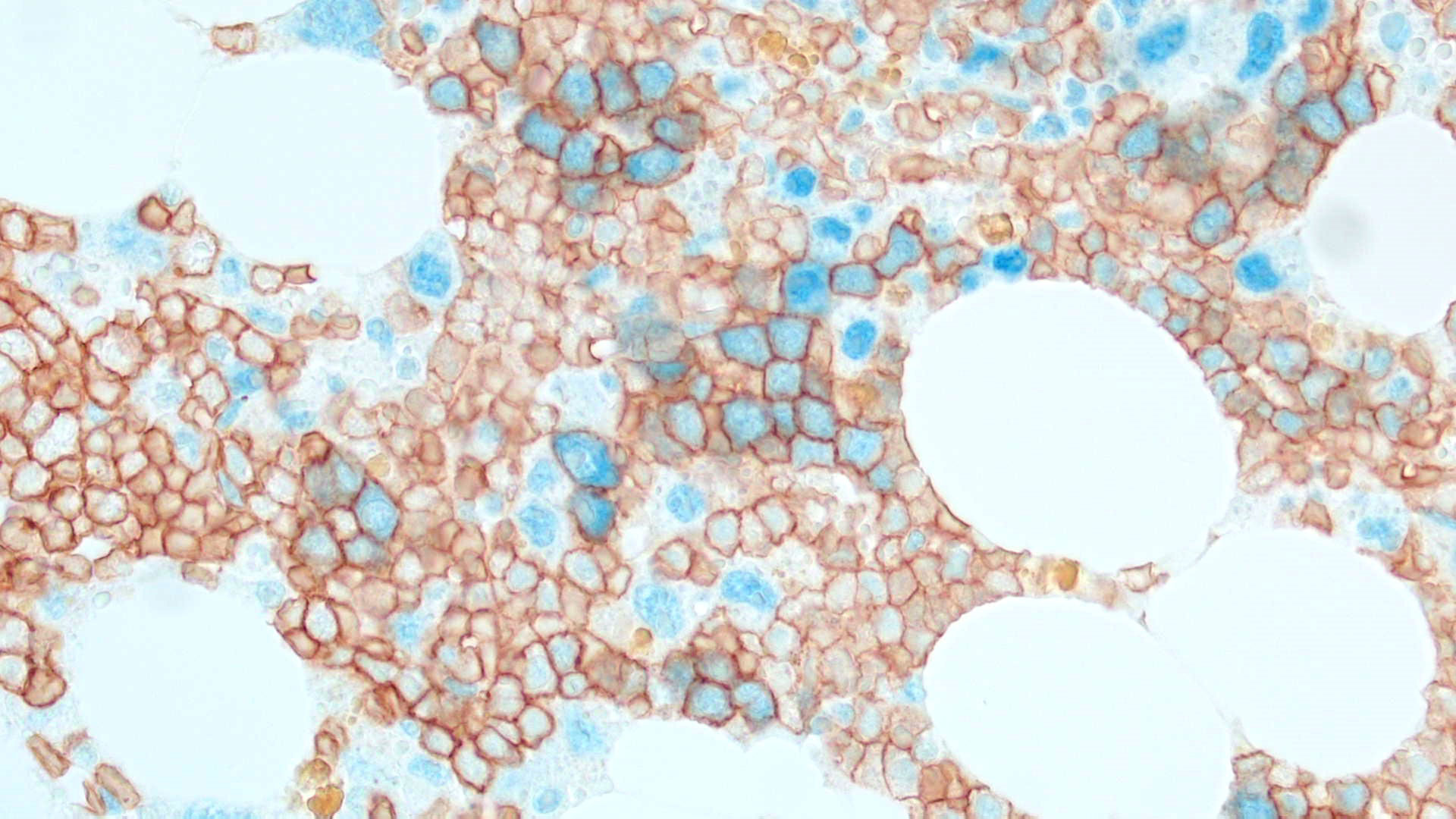
Even though about 50% of adult patients diagnosed with AML show normal cytogenetics (CN-AML), the unceasing work of Dr. Falini and other investigators has broadened the mutational landscape of the AML genome by identifying 23 recurrent mutations, including those affecting the nucleophosmin (NPM1), fms related tyrosine kinase 3 (FLT3) and DNA methyltransferase 3 alpha (DNMT3A) genes, paving the way for new targeted therapies for AML cases induced by these genetic lesions. In particular, the P.I., using immunohistochemical techniques, discovered the NPM1 mutations in 2005 and found that they represent the most frequent genetic lesions in AML (about 30% of all AML and 60% of CN-AML). The NPM1 gene encodes for a multifunctional protein with shuttling and chaperone properties that is localized in the nucleolus. The P.I. discovered that all NPM1 mutations (more than 50 so far identified) cause the same change at the C-terminus level of the NPM1 protein, i.e. the loss of the nucleolar binding domain and the generation of an additional nuclear export signal (NES) motif that are responsible for the aberrant dislocation of both mutated and wild-type NPM1 proteins in the cytoplasm of the leukemic cells. This event appears to be critical for the leukemogenic activity.
Prof. Falini played a critical role in all steps of this discovery process from the bench to the bedside. Over the past 10 years, his initial findings have been extensively validated by numerous investigators in thousands of leukemic patients, such that NPM1-mutated AML is now recognized as a disease entity in the WHO-2016 classification of lympho-hematopoietic neoplasms and evaluation of the mutational status of NPM1 (in combination with that of FLT3 gene) is now recommended as mandatory in the prognostic stratification of AML (European LeukemiaNet, 2017). Search for NPM1 mutations is also a valuable tool for monitoring of minimal residual disease in AML patients, with major impact on treatment decisions.